分子动力学模拟
一、分子动力学简介
1.1 什么是分子动力学
分子动力学(Molecular Dynamics,MD)是一种将牛顿运动定律应用于粒子系统的模拟方法。这些粒子系统的规模可以从原子、分子到颗粒部分,甚至可以是沙粒。实际上,所有的 MD 软件都遵循相同的基本步骤:
- 获取初始位置:从模拟箱中提取粒子的初始位置,并利用所选择的力场计算每个粒子所受的总力。
- 计算加速度:根据计算出的力,确定施加在每个粒子上的加速度。
- 更新速度:使用加速度来计算每个粒子的新速度。
- 更新位置:根据每个粒子的新速度和确定的时间步长,计算每个粒子的新位置。
- 循环重复:不断重复以上步骤,随着新粒子位置的更新,循环继续进行,每次处理一个非常小的时间步长。
通过这种方式,分子动力学能够模拟粒子在时间演化中的行为和相互作用。

1.2 分子动力学模拟常用软件
常用的分子动力学模拟程序有AMBER、GROMACS、OpenMM和LAMMPS等。
AMBER
Amber程序是一款用于模拟蛋白质、核酸和糖等生物大分子的分子动力学软件,包含一套完整的生物分子力场和模拟工具。Amber软件由开源的AmberTools22和商业版的Amber22组成。
GROMACS
GROMACS是一款免费开源的分子动力学模拟软件,能够高效地模拟生物系统和非生物系统的行为。它可以模拟数百到数百万个粒子的系统,包括蛋白质、脂质、核酸等复杂分子。GROMACS的开发始于20世纪90年代,最初基于GROMOS软件,后来独立发展,强调性能优化。其代码使用C语言,近年来逐步过渡到C++,并且开源。GROMACS的计算效率在多个benchmark中优于其他同类软件,因此成为生物系统分子动力学模拟领域中最常用的软件。
OpenMM
OpenMM是一款基于Python开发的开源分子动力学模拟软件。近年来,因AlphaFold的影响,该软件的热度显著提升。此外,OpenMM支持GPU硬件加速,因此在性能上表现也相当出色
LAMMPS
LAMMPS(Large-scale Atomic/Molecular Massively Parallel Simulator)是由美国Sandia国家实验室开发的开源分子动力学(MD)模拟程序,专注于材料领域的研究。它广泛应用于固态材料(如金属和半导体)、柔性物质(如生物分子和聚合物)以及粗粒度介观体系的模拟。LAMMPS内置多种原子间势(力场模型),支持对不同类型的原子和材料进行建模。
由于其灵活性和可扩展性,LAMMPS在研究者中备受欢迎,尤其是在需要进行大规模并行计算的情况下。接下来,我们将介绍如何在超算云平台使用LAMMPS软件进行分子动力学模拟。
二、在超算云平台使用lammps软件进行分子动力学模拟
2.1使用云平台lammps软件进行模拟计算
2.1.1 lammps软件安装
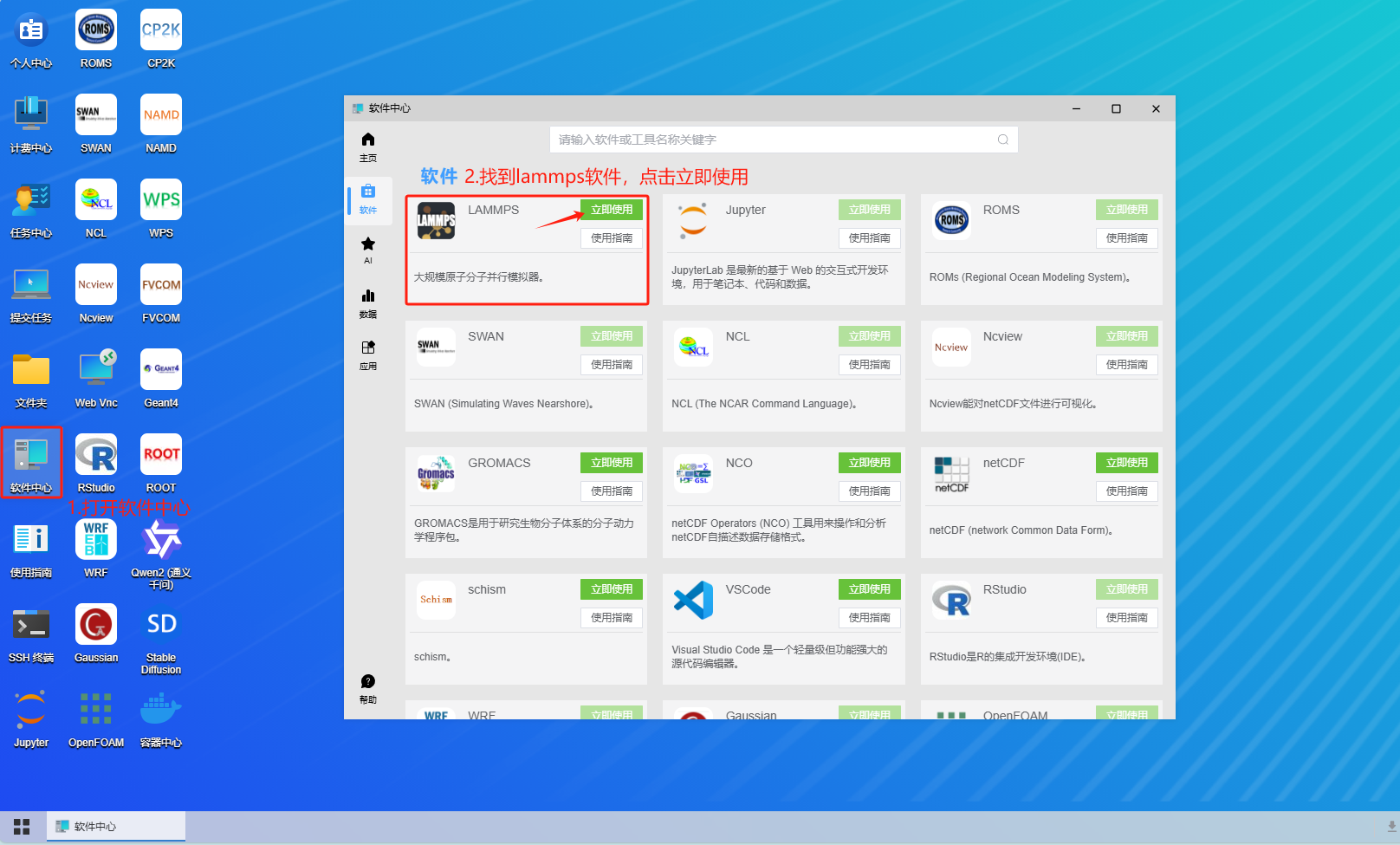
在云平台桌面打开软件中心,找到lammps软件,并点击立即使用:
2.1.2 提交作业
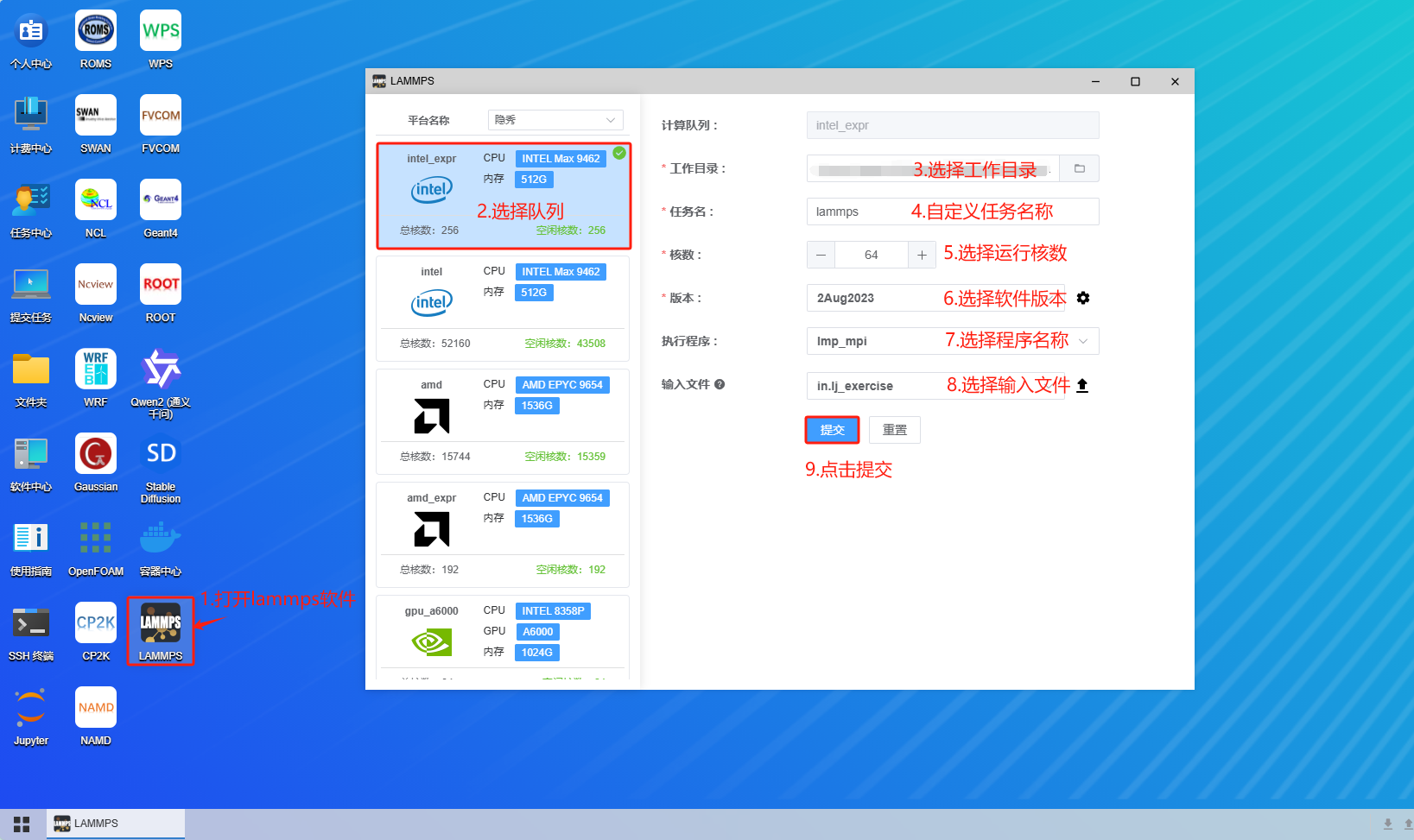
在云平台桌面选择lammps软件,选择作业提交的队列、工作目录,自定义作业名称,选择作业运行所需的核数、软件版本、程序名称以及输入文件,点击提交选项提交作业。
2.1.3 查看作业
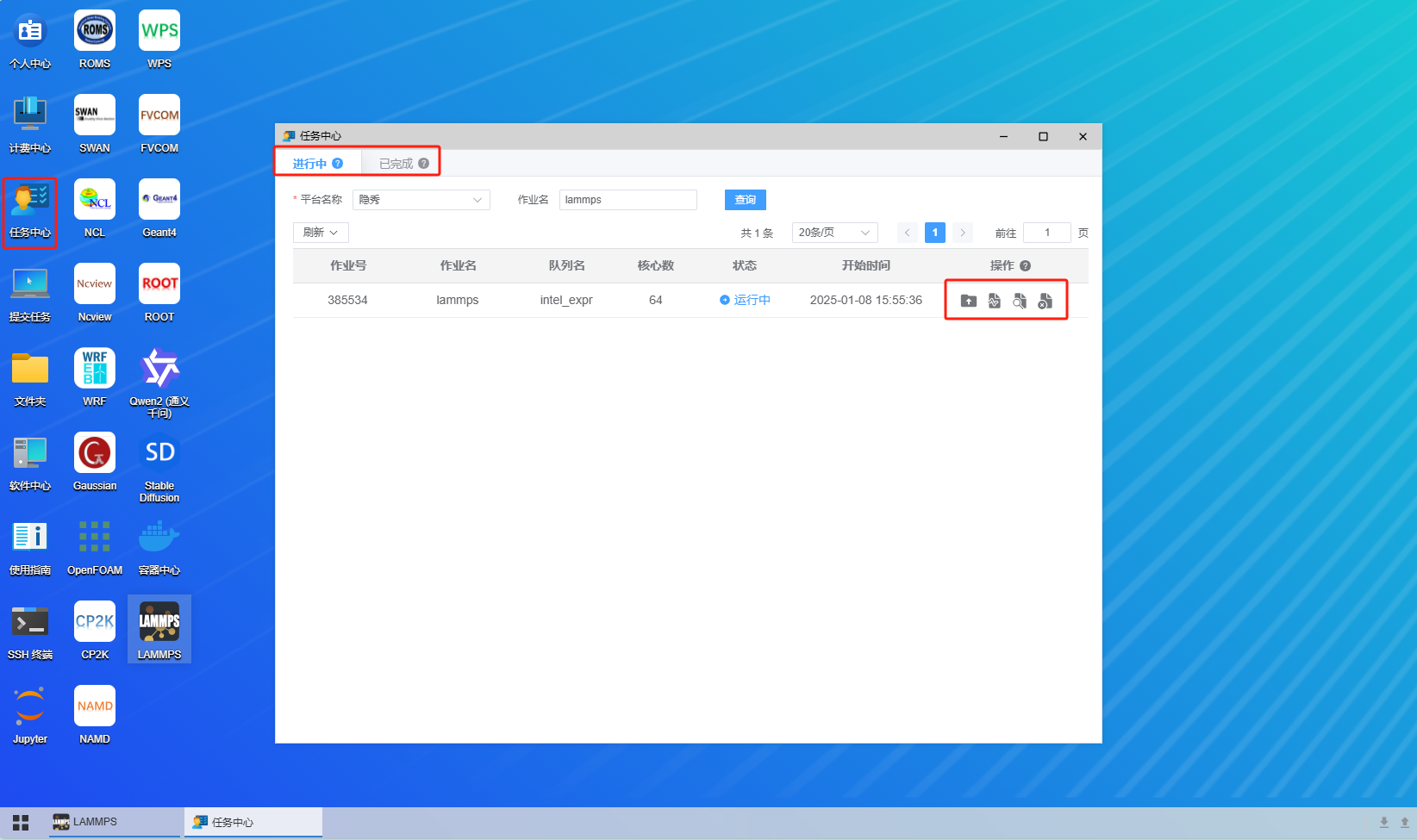
在桌面上打开任务中心, 任务中心 有两个 tab 页面:
- “进行中”:正在运行的任务,或者一分钟内结束任务
- “已完成”:已经结束的任务,一分钟同步一次
每个计算任务最右侧操作栏的四个按钮功能分别为:
- 第一个按钮:打开计算任务所在的目录
- 第二个按钮:打开程序输出日志
- 第三个按钮:打开任务详细信息
- 第四个按钮:停止计算任务
2.1.4 查看日志
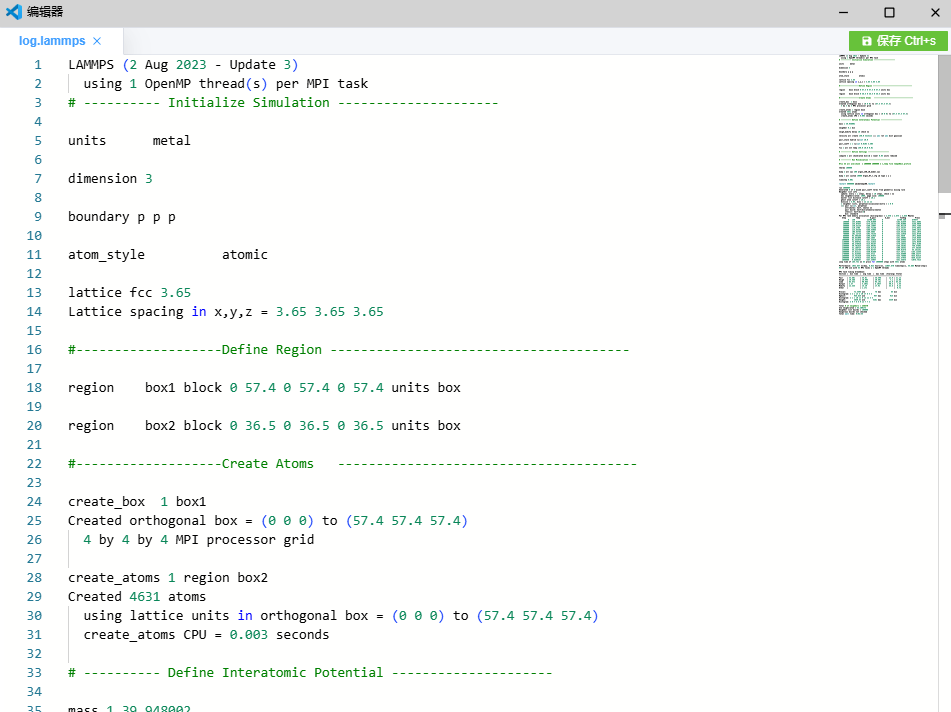
在任务中心打开作业的输出日志,查看日志内容:
2.2 使用云平台vmd软件查看分子位置信息
我们使用超算vmd软件查看算例得到的原子位置信息结果文件(.xyz文件)。
在超算云平台桌面,打开Web Vnc(vnc软件可以将计算节点图形化显示,用于使用可视化图形软件),选择想要提交的队列、工作目录,自定义任务名称、选择节点数(默认使用1个节点)、选择核心数、选择分辨率(可采用默认分辨率)、选择软件版本,点击提交,即成功提交vnc作业。
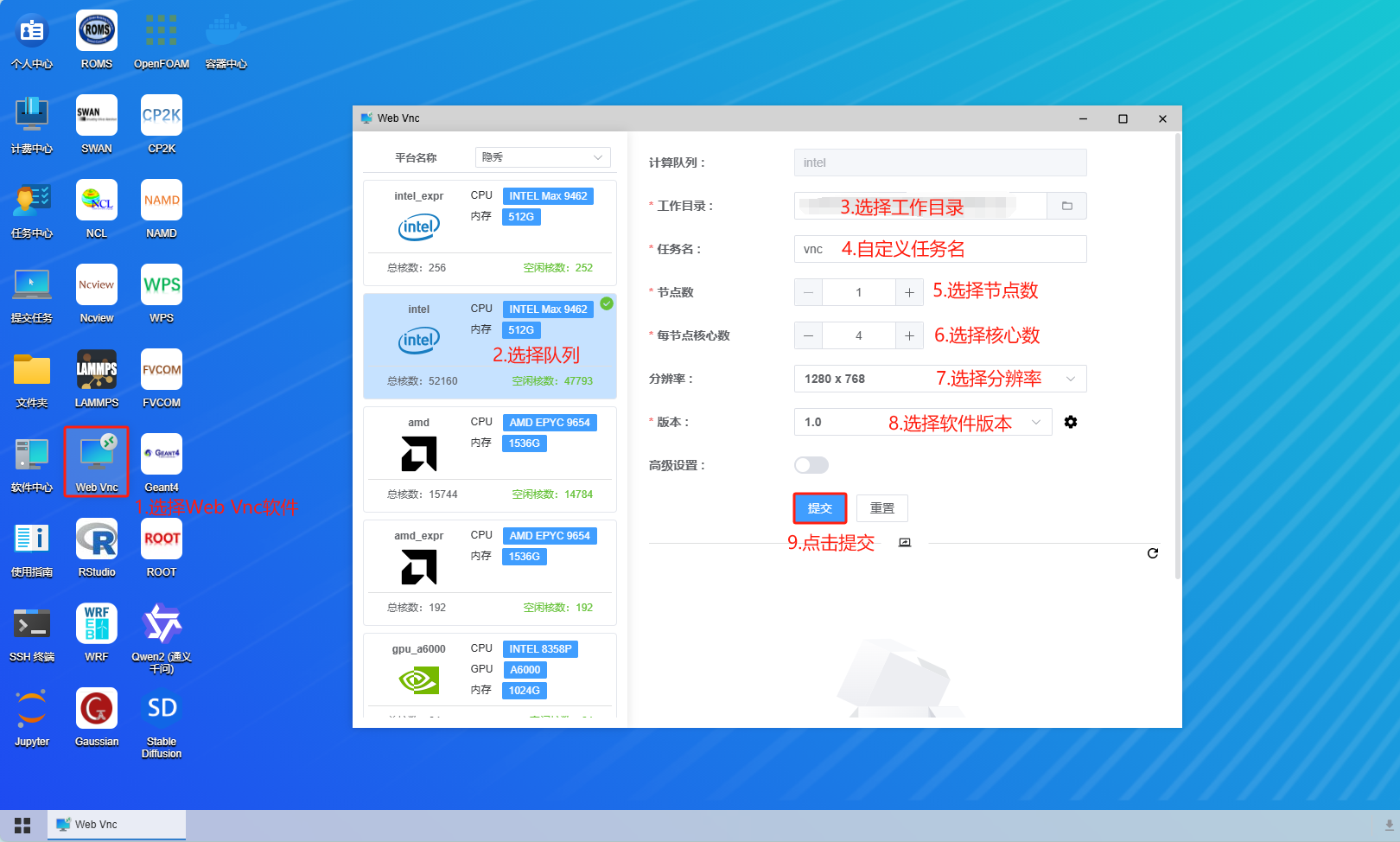
进入任务中心,打开vnc软件页面:
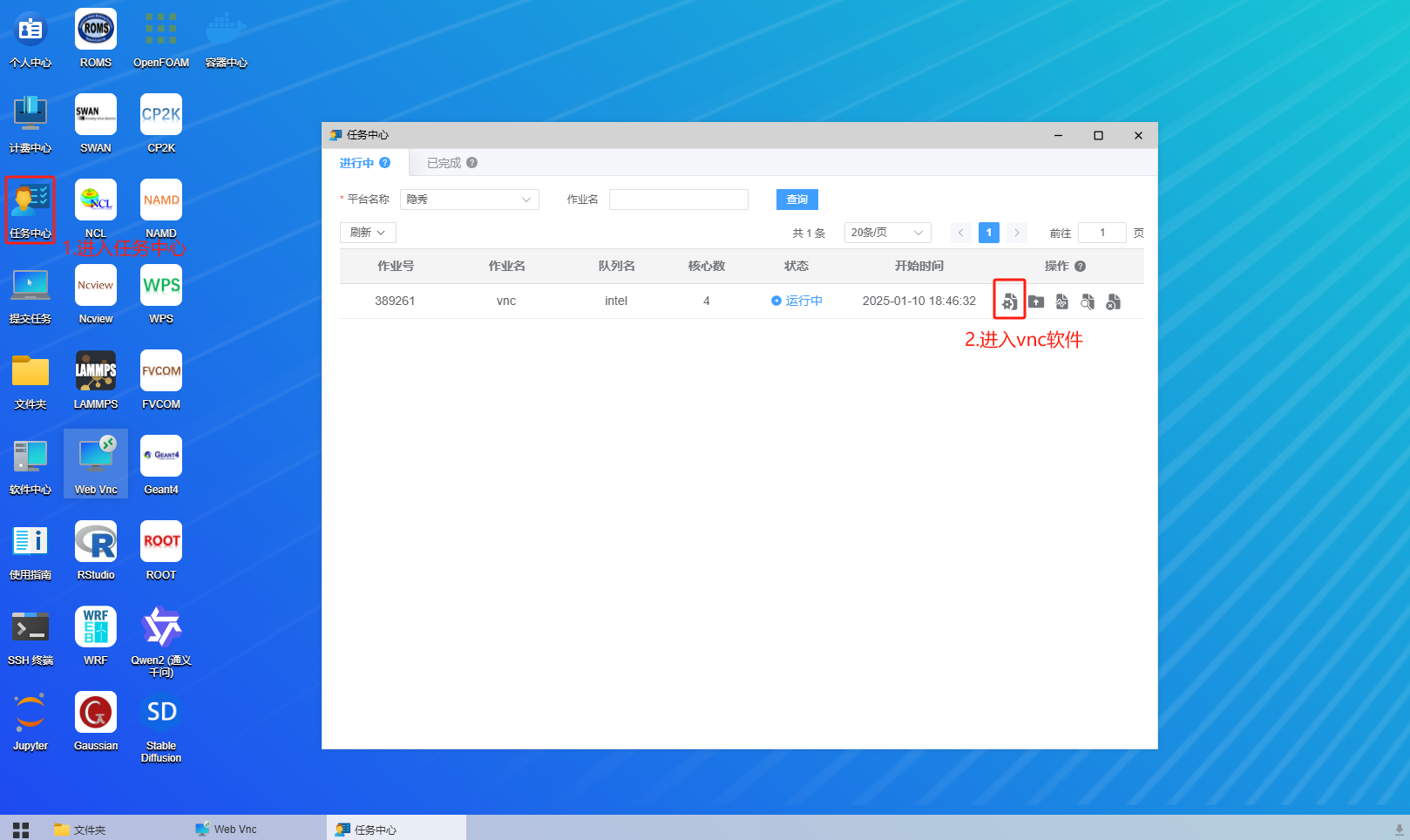
打开vnc软件后,进入终端:
依次输入以下命令,即可打开vmd软件:
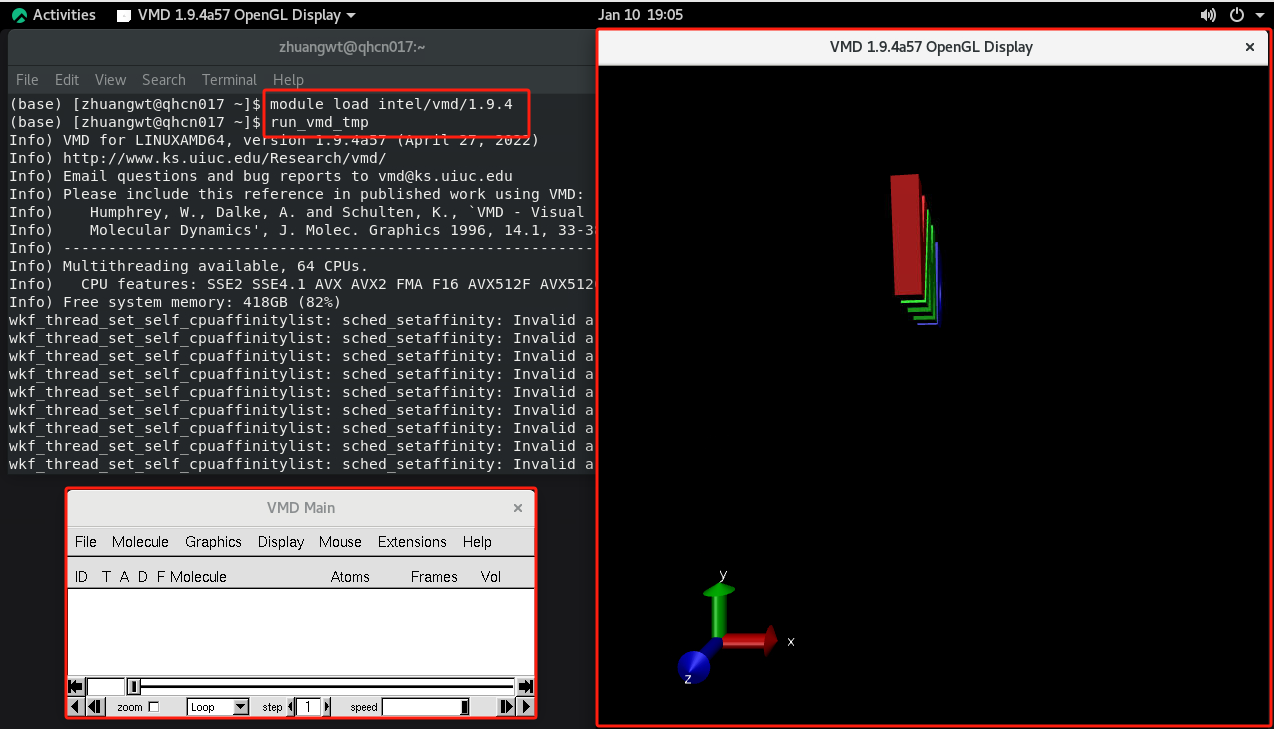
通过以下流程打开原子位置文件(.xyz文件):
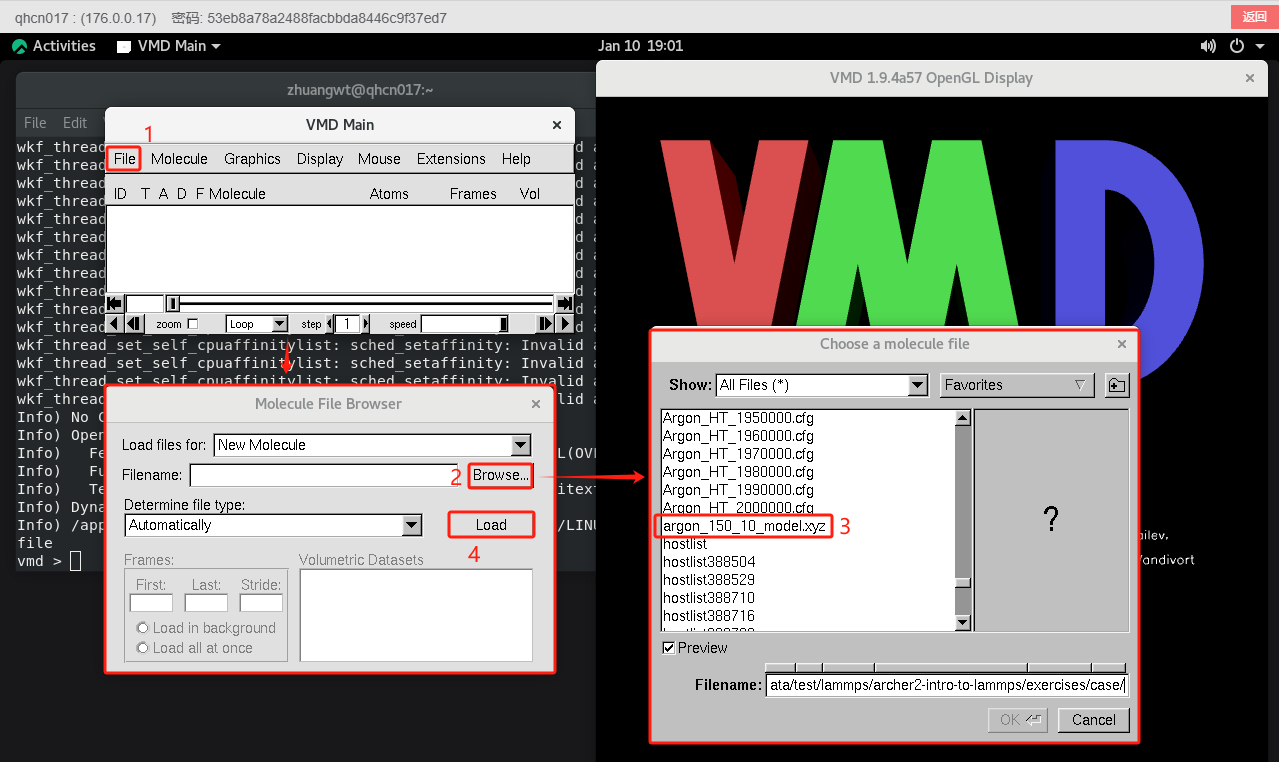
若想结束文件的查看,在关闭vnc软件的所有窗口后,在任务中心关闭vnc的任务即可。
三、使用分子动力学模拟(LAMMPS)研究纳米尺度下氩气的凝固/结晶过程
3.1 研究背景
在本案例中,我们研究纳米尺度下氩气的凝固/结晶过程,通过构建一个定义尺寸为 10 纳米左右的虚拟盒子,然后用氩原子填充。在设定了所选择的体积温度和密度后,我们在平衡状态下研究氩在盒子中的局部温度和密度分布。
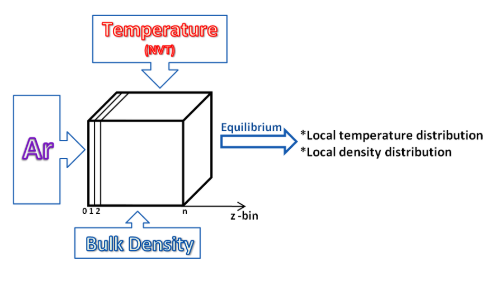
下面我们将逐步进行案例的输入文件解析、案例模拟、原子位置结果查看以及结论分析。
3.2 输入文件解析
3.2.1 仿真设置
我们要做的第一件事是选择一种单位的样式。这可以通过修改units参数值来实现:
LAMMPS 提供了多种单元样式,以满足不同类型的模拟需求。在本例中,我们选择了金属单位(metal)。金属单位是无量纲的,采用无量纲单位进行模拟计算具有明显的优势。这种选择不仅使得相关数值接近统一,还降低了对浮点变量精度的要求,从而减少了内存需求并提高了计算速度。
定义使用什么样的原子:
原子样式会影响每个原子所关联的属性,并且在模拟过程中无法更改。每种原子样式都会存储以下信息:坐标、速度、原子 ID 和原子类型。虽然原子样式不添加任何额外的属性,但其他样式可能会存储其他信息。
定义系统中的维数:
除此之外,LAMMPS 还能够模拟二维系统。
设置模拟框边界的样式:
boundary后面的三个字母分别对应三个方向(x、y、z),其中 p 表示所选边界是周期性的。除了周期性边界条件外,还可以使用其他边界类型(如固定边界、收缩包装和最小收缩包装)。
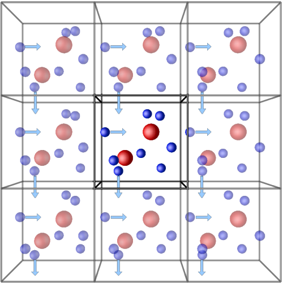
周期边界条件(PBC)允许通过模拟一个很小的单位元来近似一个无限系统。最常见的三维单元格形状是立方体,但任何可以完全镶嵌于三维空间的形状都可以使用。PBC 的拓扑结构是:当一个粒子离开单元格的一侧时,它会在另一侧重新出现。具有 PBC 的二维地图可以完美地映射到一个环面。
使用 PBC 的另一个关键方面是最小图像约定,用于计算粒子之间的相互作用。这确保了每个粒子只与其最近的粒子图像相互作用,而无论该粒子隶属于哪个单元格(无论是原始模拟框还是周期图像之一)。
定义空间中的一组点:
使用 面心立方(FCC)晶格 结构来安排原子,并且每个晶格常数为 3.65,这设定了原子的排列方式和初始位置。
命令定义空间中的几何区域:
在这个参数中,box1、box2 是我们为该区域指定的名称,block 表示区域的类型(长方体),而后面的数字分别表示 x、y 和 z 的最小值与最大值。
接下来,我们使用之前定义的区域创建一个包含一种原子类型的盒子:
最后,我们使用之前创建的盒子和格子,在盒子中生成原子,将类型为1的原子排列在上述格子的box2区域:

3.2.2 粒子间相互作用
我们已经在模拟盒子中确定了粒子的初始位置,我们还需要定义它们之间的相互作用。
定义粒子将使用的相互作用样式:
LAMMPS 提供了多种成对粒子间的相互作用。在这个例子中,我们使用的是 Lennard-Jones 相互作用,截止距离为原子直径的10倍。将相互作用截断到一定距离可以显著减少计算每个粒子能量所需的时间,因为在较大粒子间距 d 时,LJ 势能趋近于零。
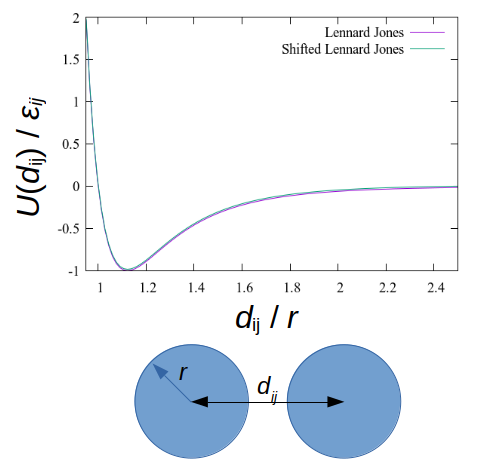
接下来,我们为原子之间的相互作用设置 LJ 参数。在这个例子中,由于只有一种类型的原子,我们只需考虑类型 1 的原子与其他类型 1 原子之间的相互作用。我们定义了两个粒子之间的最小能量 ε 和粒子直径 σ,这两者都是相对于非位移势能的。
最后,我们设定原子类型 1 (氩)的质量为 39.948002 个单位:
对于特定的模拟,可能需要许多其他特征。例如,LAMMPS 还支持模拟键合、角度、二面体和不正确角度等。
3.2.3 邻居列表
为了提高模拟性能,并考虑到我们在一定距离截断相互作用,我们可以维护一个邻近粒子的列表(在相邻的截断距离内)。这可以减少每个时间步骤所需的比较次数,这会占用少量内存。

我们可以将邻居列表的截止值设置为 0.1 σ ,因为在处理球体时,半径的小幅增加会导致体积的大幅增加。
Bin 参数是指用于构建列表的算法,对于粒子大小均匀的系统,bin 是性能最好的算法(但还有其他算法):
这些列表需要定期更新。我们使用 neigh_modify 参数设置邻居列表重建之间的等待时间:
delay 参数设置自上次邻居列表重建以来需要经过的最小时间步数,只有当时间步数等于 every 指定的值时,LAMMPS 才会尝试重建邻居列表。
如何设置邻居列表delay参数?
你可以通过运行一个快速模拟程序来估计重建邻居列表的频率,使用以下命令:
并在该运行生成的日志文件中查看结果
LAMMPS邻居列表信息然后查看生成的日志文件中的邻居列表信息。邻居列表生成将告诉你需要重新生成邻居列表的频率。如果你知道短时间模拟运行了多少时间步骤,可以通过计算每个重建平均有多少步骤来估计更新频率。如果你的更新频率小于或等于这个值,应该会看到加速效果。
3.2.4 仿真参数
现在我们已经设置了模拟的初始条件,并调整了一些设置以提高运行速度,接下来需要告诉 LAMMPS 模拟的运行方式。这包括选择集合(以及应用它的粒子)、时间步长、模拟的时间步数、输出的属性以及记录这些属性的频率。
fix 命令有多种选项,大多数与在模拟中将某些属性设置为某个值有关,或设置为某些粒子的值间隔。
第一个参数是 ID(修复的名称),第二个参数是 group-ID(应用命令的粒子)。在下面的示例中,all 表示对所有粒子应用命令:
这里的样式为 nvt,参数表示模拟开始和结束时的温度(Tstart 和 Tstop)以及温度阻尼参数(Tdam)。
3.2.5 最终设置
尽管我们在盒子中创建了许多粒子,但如果进行模拟,粒子之间不会发生任何相互作用,因为它们没有初始速度。为了解决这个问题,我们使用 velocity 参数为所选组中的粒子(在此情况下为所有粒子)生成一个速度集合:
create 样式后面的参数为温度和种子数。mom yes 表示初始设置时会保留系统的总动量,rot yes 表示会保留系统的角动量,dist gaussian 表示速度分布符合高斯分布。
接下来,我们设置时间步长的大小,单位为我们选择的模拟单位(在此情况下为 LJ 单位):
时间步长的选择是速度和准确性之间的权衡。较小的时间步长可以确保没有粒子相互作用被忽略,但会增加计算时间。较大的时间步长可以在较长时间尺度上模拟现象,但可能导致粒子在每个时间步长中移动过多,从而忽略某些相互作用——在极端情况下,粒子可能会“飞”过彼此。理想的时间步长取决于系统的类型、大小和温度,可以通过粒子的平均扩散来估计。
要设置输出频率(以时间步长为单位),可以修改 thermo 参数:
在此情况下,输出将每 100000个时间步骤打印一次。
为了输出原子位置信息,我们使用:
这将每 200 步输出一次系统的原子位置到 .xyz 格式的文件 argon_150_10_model.xyz。
下面,将每 10000 步输出一次原子的 ID、类型、坐标到文件 Argon_HT_*.cfg:
想要保存重启文件:
这会每 6000000 步保存一个重启文件,文件名为 abcdetemp100.restart。
最后,我们选择运行模拟的时间步长(而不是时间单位):
最后,我们选择运行模拟的时间步长(而不是时间单位):
3.3 提交Lammps计算
输入文件编辑完成后,保存为lmp.input文件,在超算云平台中,安装并使用lammps软件,使用该输入文件进行本案例的分子动力学模拟,详细步骤参考2.1。
3.4 氩原子位置结果文件查看
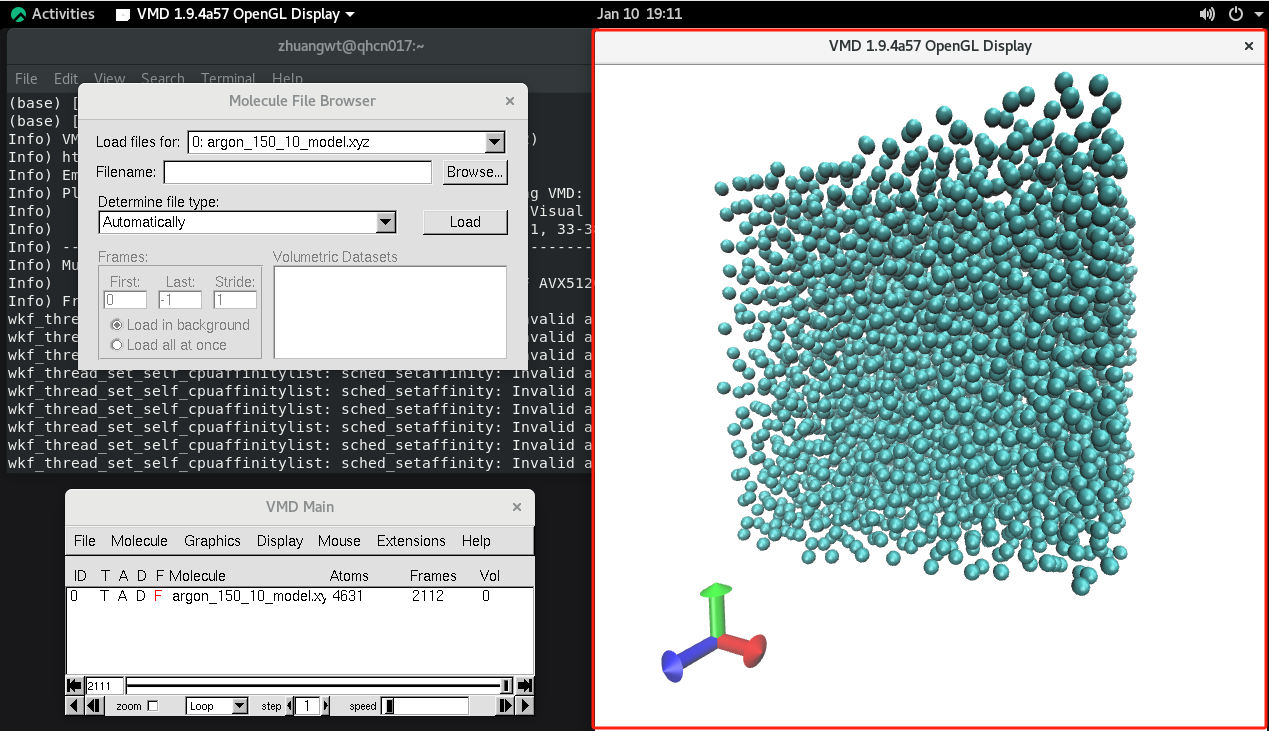
在云平台中,使用vmd软件查看分子动力学模拟后产生的氩原子位置结果文件(.xyz文件),详细步骤参考2.2,部分氩原子位置结果如下:

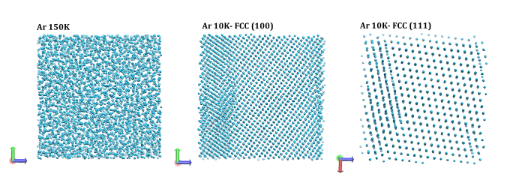
3.5 案例结论分析
对氩气 (Ar) 气相的分析表明,在气体状态下,氩原子呈现出随机分散的特征。然而,当温度降低到一定程度时,氩原子紧密结合在一起,显示出面心立方 (FCC) 结构的有序排列。